我行我show!中国医院管理案例评选,医院卓越管理实践大秀场。
点击查看
- 具有表皮生长因子受体(EGFR)外显子 19 缺失或外显子 21(L858R)置换突变的局部晚期或转移性非小细胞肺癌(NSCLC)成人患者的一线治疗;
- 既往经 EGFR 酪氨酸激酶抑制剂(TKI)治疗时或治疗后出现疾病进展,并且经检测确认存在 EGFR T790M 突变阳性的局部晚期或转移性 NSCLC 成人患者的治疗;
- 作为首个辅助疗法(adjuvant therapy),治疗肿瘤携带特定类型基因突变的非小细胞肺癌(NSCLC)患者。
2020年销售额43.28亿美元,折合人民币283亿元。
今天笔者跟大家分享一下奥希替尼的研发历程。

EGFR是一组具有酪氨酸激酶活性的表皮生长因子家族的细胞表面受体,由一个细胞外配体结合区、一个跨膜区和一个具有酪氨酸激酶结构域的细胞内区域组成。
当生长因子(如表皮生长因子EGF)与胞外结构域结合时,该受体将与其自身或其他家族成员发生二聚化,诱导三磷酸腺苷(ATP)介导的自磷酸化,从而启动激活下游多条信号传导通路(包括PI3K-Akt、MAPK-Erk等),产生细胞增殖效应。
如果EGFR活化异常,可持续激活与肿瘤增殖、分化相关的基因,继而诱发肿瘤的形成和发展。
EGFR在非小细胞肺癌NSCLC患者突变类型中突变率最高,EGFR已经成为非小细胞肺癌治疗的主要靶点之一。

20世纪90年代,阿斯利康开启了一系列的创造性研究,发现了苯胺喹唑啉类EGFR抑制剂吉非替尼(商品名:易瑞沙),吉非替尼是全球第一个上市的EGFR抑制剂。
之后,相继出现吉非替尼的“me-too”药物厄洛替尼、埃克替尼。

上面已经提到,EGFR活化异常,可持续激活与肿瘤增殖、分化相关的基因,继而诱发肿瘤的形成和发展,研究发现,肿瘤细胞EGFR激活突变位点位于不同于活性位点的激酶结构域中。其中一个激活突变位点位于第858位的亮氨酸(Leu,简称L),通过点突变为精氨酸(Arg,简称R),这一突变点形式描述为“L858R”;另外一个激活突变位点是746~750的残基缺失,对应的是EGFR基因外显子19(exon19 del)。
EGFR激酶结构域与易瑞沙复合体的晶体结构
“L858R”和“exon19 del”能够持续激活EGFR,最终导致肿瘤细胞的不断增殖、分化。
吉非替尼等抑制剂对EGFR激活突变体的亲和力相比野生型(正常非突变EGFR)更强,吉非替尼对突变型L858R的Kd值=2.4nmol/L,而对野生型的Kd值=35nmol/L,两者相差15倍。(注:解离常数Kd=一半的受体被配体结合时,配体的浓度,反映出化合物对靶标的亲和力,值越小,亲和力越强)。
由此可知,携带上述两种突变EGFR的肿瘤细胞对EGFR抑制剂的敏感性远远高于携带野生型EGFR的正常细胞。
同时发现,携带上述突变的EGFR,与ATP(介导EGFR的正常激活)的亲和力降低,这就会导致携带上述突变EGFR的肿瘤细胞,在正常ATP浓度范围内,在吉非替尼等抑制剂作用下,其更容易被更好的抑制激活。
以上便是吉非替尼等抑制剂对EGFR激活突变肿瘤抑制作用的分子理论基础。

但是令人遗憾的是,上述抑制剂10个月左右的时间便会发生耐药,超过半数的耐药原因是肿瘤细胞EGFR激酶结构域的又一新型突变,这个突变就是现在认识比较清晰的790位苏氨酸(Thr,简称T)点突变为甲硫氨酸(Met,简称M),一般称之为“T790M”。
现在也就有了“L858R”合并“T790M”、“exon19 del”合并“T790M”的新型双突变组合,这是一个全新的挑战,这一问题足以使得第一代诞生的吉非替尼等抑制剂失去作用,肿瘤细胞逃脱抑制疯狂生长。
因为吉非替尼对双突变体的Kd值,与野生型相比已经没有优势可言(11nmol/L VS 35nmol/L),同时双突变体EGFR对ATP(介导EGFR的正常激活)的亲和力(Km值)与野生型EGFR相比基本相当(8.4μmol/L VS 3.5μmol/L)。
体外细胞实验结果也提示,吉非替尼对携带双突变EGFR的肿瘤细胞已经没有明显的选择性抑制作用。

阿斯利康科学家设计的第一代EGFR-TKI吉非替尼出现不久,基于吉非替尼苯胺喹唑啉骨架而设计出的第二代EGFR-TKI应运而生。
第二代EGFR-TKI代表药物有勃林格殷格翰的阿法替尼、辉瑞制药的达克替尼,这一类药物的最大结构特点是在苯胺喹唑啉的基础之上,引入了迈克尔加成受体丙烯酰胺。

之所以采取上述结构修饰策略,主要是考虑到EGFR激酶配体结合位点含有半胱氨酸残基(C797),迈克尔加成受体能够与该残基形成共价结合。
EGFR激酶T790M与阿法替尼复合物的晶体结构
研究人员推断,第一代可逆EGFR-TKI出现的耐药问题,部分原因是ATP亲和力出现变化,如果采用不可逆抑制剂,有可能解决这一问题。
然而,细胞实验研究表明,阿法替尼、克唑替尼等对双突变EGFR肿瘤细胞和野生型EGFR的正常细胞没有选择性(抑制活性为23nmol/L VS 12nmol/L)。
“L858R”合并“T790M”、“exon19 del”合并“T790M”的双突变组合依然无法克服。

双突变“L858R”合并“T790M”的EGFR,构成肿瘤细胞疯狂生长的利剑。
药物化学家猜测,在EGFR激酶中,看门氨基酸的性质对抑制剂的活性可能发挥重要作用,他们不断将目光转向靶向甲硫氨酸看门氨基酸残基的化合物上面。
基于此,很快合成了一组复合预期设计目标的化合物,并进行了活性筛选试验。
活性筛选中,研究人员发现了关键的苯胺嘧啶类化合物1,化合物1对双突变EGFR表现出较高的抑制活性(IC50=9nmol/L)和优良的选择性。
然而,令人遗憾的是,化合物1在H1975细胞中表现出对EGFR自磷酸化较低的抑制活性,而且大量合成化合物1的类似物面临同样的尴尬问题。
药物化学家猜测可能的原因是,双突变体EGFR的ATP亲和力增加,底物和抑制剂竞争ATP结合导致。
将第一代EGFR-TKI设计成不可逆共价结合(靶向797位半胱氨酸残基)的阿法替尼策略,运用到解决现在的问题,研究人员进行了尝试,成功设计并合成出化合物2。

果不其然,化合物2对“L858R”合并“T790M”、“exon19 del”合并“T790M”的EGFR肿瘤细胞均表现出明显的抑制活性,同时具备一定的选择性。
化合物2与EGFR复合物晶体结构
不过化合物2的药代性能不佳,药物化学家基于此,设计合成系列化合物,并最终筛选出满足预期目标的化合物3。
之后研究人员不断进行摸索修饰,得到化合物4、5。
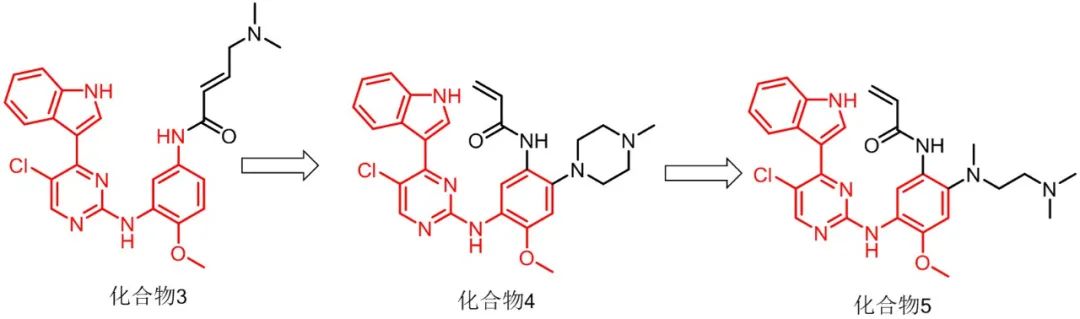
但是药物复杂性远远不是上述的定向设计这么简单,药物化学家发现化合物5可能面临着严重副作用,这是因为其能够抑制IGF1R,而IGF1R与胰岛素受体酪氨酸激酶同源性较高,最终可能导致化合物5引起血糖紊乱的问题,除此之外化合物5还有其他毒副作用被发现。
即便化合物5有诸多不完美,但是它距离最终发现药物化学家心目中理想的EGFR-TKI已经不远了。
以化合物5为先导化合物,对其进行进一步修饰改造,除了毒副作用需要重点考虑之外,结构改造还需要注重提高化合物亲脂性配体效率方面(LLE)。
化合物6、7、8和9先后被设计并合成,最终优选出化合物9,化合物9不仅药动性能优良,其对IGF1R的活性也有了显著的降低,这一点相比其他化合物的优势独一无二。

化合物9还有另一个优势,就是其对双突变EGFR优良的选择性,对于其他大多数已知激酶,化合物9的抑制作用并不明显。
通过质谱分析,证实化合物9与野性型EGFR发生共价结合。
化合物9与野生型EGFR结合(PDB:
4ZAU)
又经过一段小插曲,在此不再赘述,最终化合物9确定为候选临床药物,命名AZD9291(Osimertinib)。
2015年11月,FDA加速批准了阿斯利康的Osimertinib(商品名:TAGRISSO)用于治疗正在或已经接受EGFR激酶抑制剂治疗且呈转移性EGFR T790M突变阳性的NSCLC患者。
2018年4月19日,FDA批准奥希替尼作为EGFR突变(外显子19缺失或外显子21 L858R突变)的转移性非小细胞肺癌(NSCLC)患者的一线治疗药物。
2020年12月19日,美国FDA正式宣布奥希替尼作为首个辅助疗法(adjuvant therapy),治疗肿瘤携带特定类型基因突变的非小细胞肺癌(NSCLC)患者。
参考:
1.《成功药物研发Ⅰ》[匈] 亚诺斯·费舍尔等 著,白仁仁 译
2.阿斯利康2020年业绩公告.
3.艾力斯上市招股书.
4.http://www.rcsb.org/structure/2ITY.
EGFR,奥希替尼,吉非替尼,抑制剂,化合物,药物














 打赏
打赏




















 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612





 京公网安备 11010802020745号
京公网安备 11010802020745号
