EGFR激酶激活突变存在于
10–15%的白种人和50%的东亚非小细胞肺癌患者中,这些激活突变导致组成性配体非依赖性受体激活并促进细胞存活和增殖。EGFR激酶19外显子缺失和21外显子L858R点突变对EGFR TKIs治疗敏感。

几个III期临床试验研究结果证实,与顺铂对比:第一代
(吉非替尼和厄洛替尼)
和第二代
(阿法替尼他尼和达科替尼)
EGFR TKIs的中位缓解率为70-75%,无进展生存期
(PFS)
在10-14个月;而第三代EGFR-TKI-奥希替尼显示出更优越的疗效,显著改善了PFS
(18.9对10.2个月)
和总生存率
(38.6对31.8个月)
。尽管疗效突显著,但耐药不可避免地出现,并导致疾病进展。
在EGFR突变阳性患者中,TKI治疗的选择性压力可能导致敏感克隆的消除,获得靶标依赖性和非依赖心耐受机制,耐药克隆的存在和诱导适应性耐药的出现决定了EGFR TKI耐药的时间,并表现出先天性或获得性耐药。耐药机制包括靶蛋白突变耐药,非靶标依赖性信号通路扩增、激活及表型转化等
(图1)
。图1 | EGFR信号转导通路,获得性耐药机制和潜在治疗策略不同EGFR抑制剂的靶标依赖性(on-target, EGFR-dependent)和非依赖性(off-target, EGFR-independent)耐药机制有很大不同。接受第一代或第二代EGFR-TKIs的患者主要产生EGFR依赖性耐药,而接受第三代TKI奥希替尼作为二线治疗的患者中只有约20%表现为EGFR依赖性耐药机制。在一线奥西替尼治疗的患者中,约10-15%的患者表现为EGFR依赖性耐药(图2)。
图2 |奥西替尼的耐药机制
a、 二线(a)和一线(b)奥西美替尼治疗的耐药机制
T790M突变:在接受吉非替尼、厄洛替尼或阿法替尼治疗的患者中,50-60%的患者发现T790M突变
(图3)
耐药,但对三代EGFR抑制剂奥希替尼敏感。有趣的是,治疗前T790M突变的存在已被广泛报道,其发生率变化很大
(<1–65%)
,并且可能与临床不良预后有关。在III期随机AURA3试验中,与以铂为基础的化疗相比,奥希替尼显著延长PFS
(中位PFS=10.1 vs4.4个月)
,对脑转移患者同样有效
(中位PFS=8.5 vs 4.1个月)
。虽然奥西替尼有更高的应答率
(71对31%)
,但在这项研究中,并没有统计上显著的总生存获益
(中位总生存期=26.8对22.5个月)
。随机III期AURA3试验中接受二线奥西美替尼治疗的T790M阳性患者的血浆基因分型数据显示,约50%的患者在疾病进展时保留了T790M突变;其余患者在进展为奥西美替尼时T790M突变缺失,与早期耐药和较短生存期相关。C797X突变
:Cys797是奥希替尼共价结合的位点,C797X突变已成为最常见奥西美替尼耐药机制。丝氨酸是最常被取代的氨基酸
(p.Cys797Ser)
,而甘氨酸
(p.Cys797Gly)
也有报道。在AURA3试验中,
奥
西美替
尼
二线
治疗患者的C797x突变几率为
15%;而
T790M阳性患者
在接受第三代EGFR TKIs治疗时突变率较高,达到
22–25%。
在奥西替尼一线治疗患者中,C797X突变的发生率较低,
FLAURA试验中79名患者的发生率
只有7%
。两个方面对C797X介导的耐药性的管理至关重要。首先,在缺乏T790M突变的情况下,仅有C797S突变的NSCLC可能对喹唑啉类EGFR抑制剂
(包括吉非替尼、厄洛替尼和阿法汀)
保持敏感性。其次,当C797S和T790M突变为反式时,肿瘤对第一代和第二代EGFR TKIs仍保持敏感性;当这些突变发生在顺式位置时,则无效。临床资料显示,66%的病例有顺式表达。其他EGFR依赖性耐药机制:
EGFR第20外显子的溶剂前沿突变
G796R、G796S和G796D
,涉及铰链区的
L792X突变
等可能干扰奥希替尼与与靶蛋白的结合。其他罕见的EGFR TKI耐药突变,如EGFR第
18
外显子
ATP结合位点的L718和G719残基,以及激酶P环域的G724S突变。
据报道,在没有T790M突变的情况下,这些突变对第一代和第二代EGFR TKIs保持体外敏感性。也有报道发现,部分病人在第三代EGFR-TKIs(奥希替尼和rociletinib)治疗失败后,存在野生型EGFR基因扩增。
图3 | osimertinib治疗后获得性EGFR依赖性突变
MET扩增:
无论使用何种EGFR-TKI或何种治疗方法,MET受体酪氨酸激酶信号通路是EGFR治疗后参与EGFR抵抗的最常见的信号途径,下游信号包括STAT、MAPK、PI3K等。
目前,MET扩增最广泛采用的定义是MET基因拷贝数的存在≥5或MET/CEP7比值≥2
,但免疫组化法检测MET过表达
也已在进行中的临床试验中采用
。
在接受第一代EGFR TKIs治疗后,
5-22%的患者出现MET扩增
。奥希替尼二线和一线治疗的患者中,MET扩增率分别为10-19%和15%。但在未经选择的EGFR突变晚期NSCLC患者中,与单独使用EGFR-TKI相比,联合靶向EGFR和MET的第一代EGFR-TKI和抗MET单克隆抗体的策略没有显示出生存优势。HER2扩增
:HER2基因编码ErbB2受体酪氨酸激酶,通过MAPK和PI3K通路的交替激活介导EGFR-TKI抗性。在无T790M突变接受第一代EGFR TKIs的患者中,
12%的肿瘤样本中检测到HER2扩增
。奥西替尼耐药性的的血浆基因分型数据表明,
AU
RA3试
验的
二线患者
中,
5%的患者HER2扩增;
在FLAURA研究的一线患者中,2%的患者HER2扩增。
有趣的是,在所有观察到的发现中,HER2拷贝数增加/扩增和EGFRT790M突变是相互排斥的。致癌融合/染色体重排:
在4-7%的患者中发现了几种涉及驱动癌基因的基因融合,主要与奥希替尼的二线治疗耐药机制有关,包括RET
(RET–ERC1,CCDC6–RET和NCOA4–RET)
,BRAF
(AGK–BRAF,ESYT2–BRAF,PCBP2–BRAF和BAIAP2L1–BRAF)
,NTRK
(TPM3–NTRK1)
,ROS1
(GOPC–ROS1)
和FGFR
(FGFR3–TACC3)
等。在一线osimer-tinib治疗中,报道了一例ALK融合
(SPTBN1–ALK)
病例。其他机制:
FLAURA试验的第一代TKI治疗队列中1%的患者
具有NRAS突变;
在第一代TKIs治疗进展的患者中,二线奥希替尼治疗后发现了可变KRAS突变,有3%的患者观察到BRAFV600E突变。临床试验中发现的其他异常包括PIK3CA突变、PTEN缺失
,CDK4、CDK6、CDKN2A的扩增或突变等;
体外研究中发现罕见的非EGFR依赖耐药机制包括FGFR、Src、AXL等信号改变。针对这些耐药机制,不同的临床试验正在进行之中,试图解决上述耐药。
组织学和表型转换:
在EGFR突变型肺癌中,有
14%的人在第一代EGFR TKIs后转化为小细胞肺癌(SCLC)
。在第三代EGFR TKIs上发生疾病进展的患者SCLC转化的比例相似(4–15%),无论是一线还是二线治疗。
EGFR的基础突变状态与SCLC转换时间有关,RB1和TP53基因也与表型转化有关。
18%
存在的RB1和TP53突变的患者
发生SCLC转化,而野生型RB1和TP53的EGFR突变阳性患者中没有SCLC病例。
这类患者的预后较差
,尽管有报道称铂和依托泊西德有初步反应,但常规全身化疗疗效有限。鳞状细胞转化也是最近发现的一种获得性EGFR-TKI耐药机制。特异性抗性机制的鉴定导致生物标记物驱动疗法的发展。目前正在评估靶向多个耐药节点的组合方法
(图1)
以及预先阻止选择耐药克隆的靶向策略
(表1-3)
。生物标志物驱
动的策略。
第四代EGFR抑制物
(克服C797S和T790M突变)
如EAI045、JBJ-04-125-02和BLU-945已单独或与奥西美替尼联合进行体外和体内活性测定,但尚未达到临床试验阶段。在C797X存在和T790M缺失的情况下,奥希替尼失败后使用第一代或第二代EGFR TKIs,以及奥希替尼与前一代TKIs结合用于C797X和T790M的反式表达已经被报道。有趣的是,除第四代EGFR-TKI外,ALK抑制剂brigatinib对三重突变EGFR
(敏感突变/T790M/C797S)
具有体内活性。针对MET扩增耐药,
进行中的联合实验方案包括:1)奥西莫替尼+MET-TKI-savolitinib;2)
capmatinib(一种MET抑制剂)+第一代
EGFR-TKI-吉非替尼;3)
MET-TKI-tepotinib+吉非替尼;4)
双特异性EGFR和c-MET单克隆抗体amivantamab与第三代EGFR-TKI-lazertinib联合等
。针对HER2扩增耐药性,进行中的联合实验方案包括奥希替尼+抗人表皮生长因子受体2(HER2)抗体-药物缀合物(ADC)trastu-zumab-emtansine(T-DM1)等。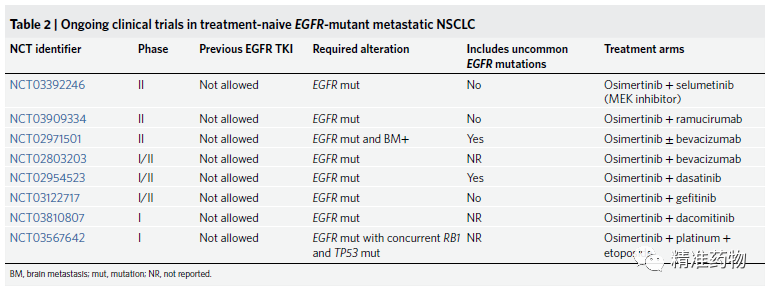
不可知论策略
。
在缺乏特异性耐药机制的情况下,生物标记物驱动的方法是不可行的。在这些情况下经常使用化疗,因为EGFR突变的NSCLC患者对铂类化疗敏感。化疗期间继续给予EGFR TKI仍有争议。例如,在一线吉非替尼治疗后的III期临床试验中,继续使用吉非替尼的患者的PFS和总体生存率没有差异。在EGFR突变阳性疾病中,免疫检查点抑制剂单一疗法并未显示出优于化疗,而免疫检查点抑制剂联合化疗仅在与抗血管生成药物相关时显示出益处。由于HER3在EGFR突变肿瘤中经常被过度表达,
一种新的HER3导向
ADC药物
patritumab-deruxteca在EGFR-TKI预处理的患者中显示出良好的结果
。
有趣的是,无论是否存在EGFRC797S突变、MET扩增、HER2突变、BRAF融合或PIK3CA突变(NCT03260491),都可以观察到疗效。TROP2是一种细胞内钙信号转导子,在非小细胞肺癌中常过表达。DS-1062a的一期研究(NCT03401385)
显示了
令人鼓舞的结果。防止耐药的出现
。
延缓EGFR-TKI耐药性的主要非选择性方法是联合应用EGFR-TKIs和化疗。
先前在未选择EGFR突变状态的NSCLC患者中,将第一代EGFR TKIs与化疗相结合,与单独化疗127–130相比,无论EGFR突变状态如何131,都没有额外的生存益处。然而,在选定的EGFR突变患者中,吉非替尼与卡铂联合培美曲塞与NEJ005二期试验中,联合组的PFS显著延长
(20.9个月比11.9个月;HR=0.490;P<0.001)
,同时观察到总体生存优势。EGFR-TKIs与抗血管生成药物的联合应用已经在一线进行了研究,目的是延缓耐药性,尽管临床上VEGF/VEGF受体抑制如何增强EGFR抑制剂作用的机制基础尚不清楚。厄洛替尼和抗VEGF单克隆抗体贝伐单抗的联合应用在单臂II期试验中显示出显著的PFS益处。但奥希替尼加
贝伐单抗
临床实验显示
,
与单独使用奥西美替尼相比,
联合治疗对T790M阳性患者的PFS没有延长。在确诊时,EGFR突变NSCL患者的脑转移发生率为24-32%。据报道,经第一代EGFR TKIS治疗的EGFR突变阳性NSCLC患者,2年累积CNS转移发生率为20%,在已有CNS疾病的患者中该值达到47%。第一代和第二代EGFR TKIs对脑转移显示出不同的活性,厄洛替尼显示出更好的CNS数据。特别是,在早期临床试验中,高脉冲剂量的厄洛替尼可以增加脑脊液中的药物浓度,产生高颅内反应率
(75%)
。第三代奥西美替尼在临床前模型中表现出CNS活性。包括
128例患者的
AURA-Extension和AURA2二期临床试验结果表明,
在50名可评估的患者中,无论是否接受过脑放射治疗,ORR为54%,疾病控制率为92%。
迄今为止,无论是否累及中枢神经系统,一线奥西美替尼是EGFR阳性晚期非小细胞肺癌的标准治疗方案。
奥希替尼对中枢神经系统疾病的活性可以减少或延缓中枢神经系统的进展。尽管在实验环境中解决治疗耐药性方面取得了显著进展,但以铂类药物为基础的化疗是迄今为止唯一获批的治疗奥西替尼替尼治疗后进展患者的临床方案。










































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612
 京公网安备 11010802020745号
京公网安备 11010802020745号










